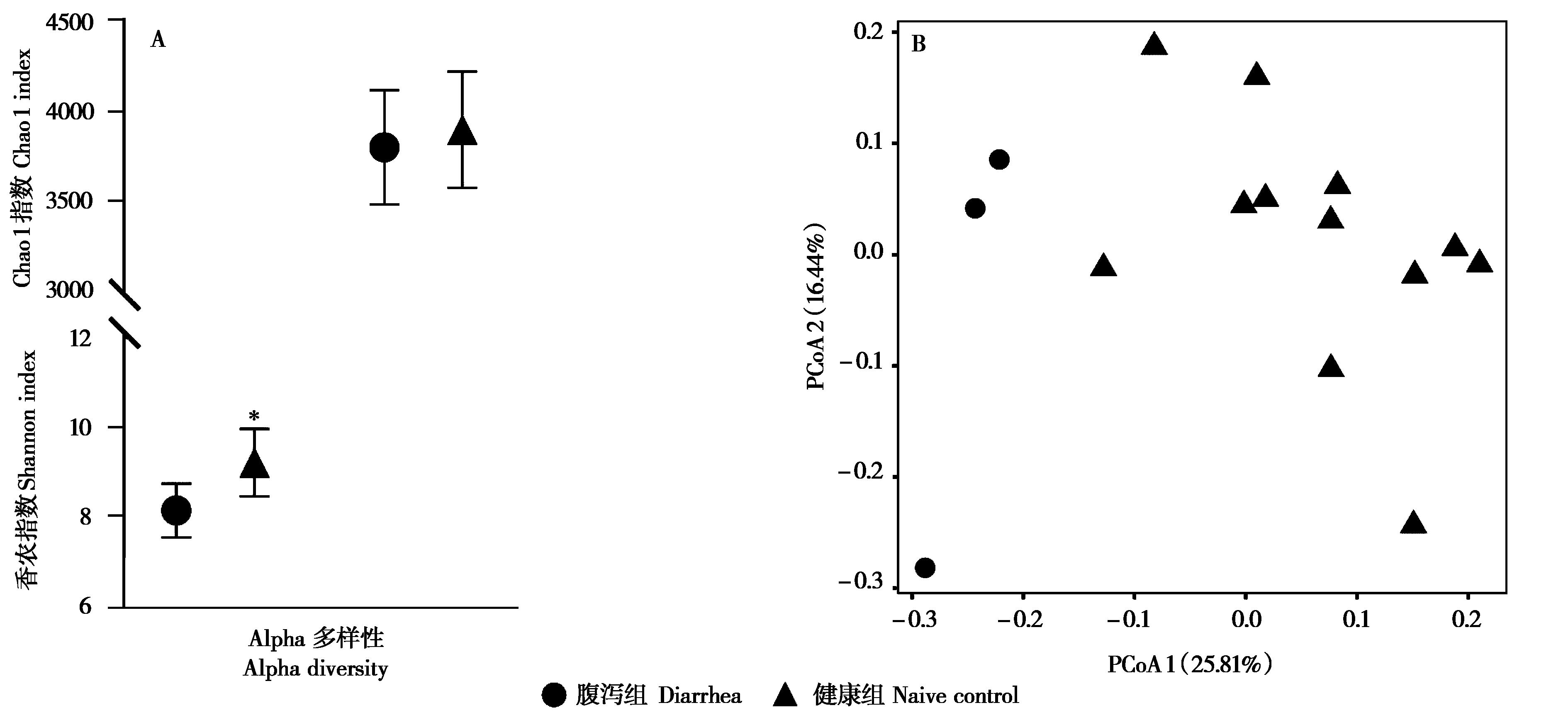
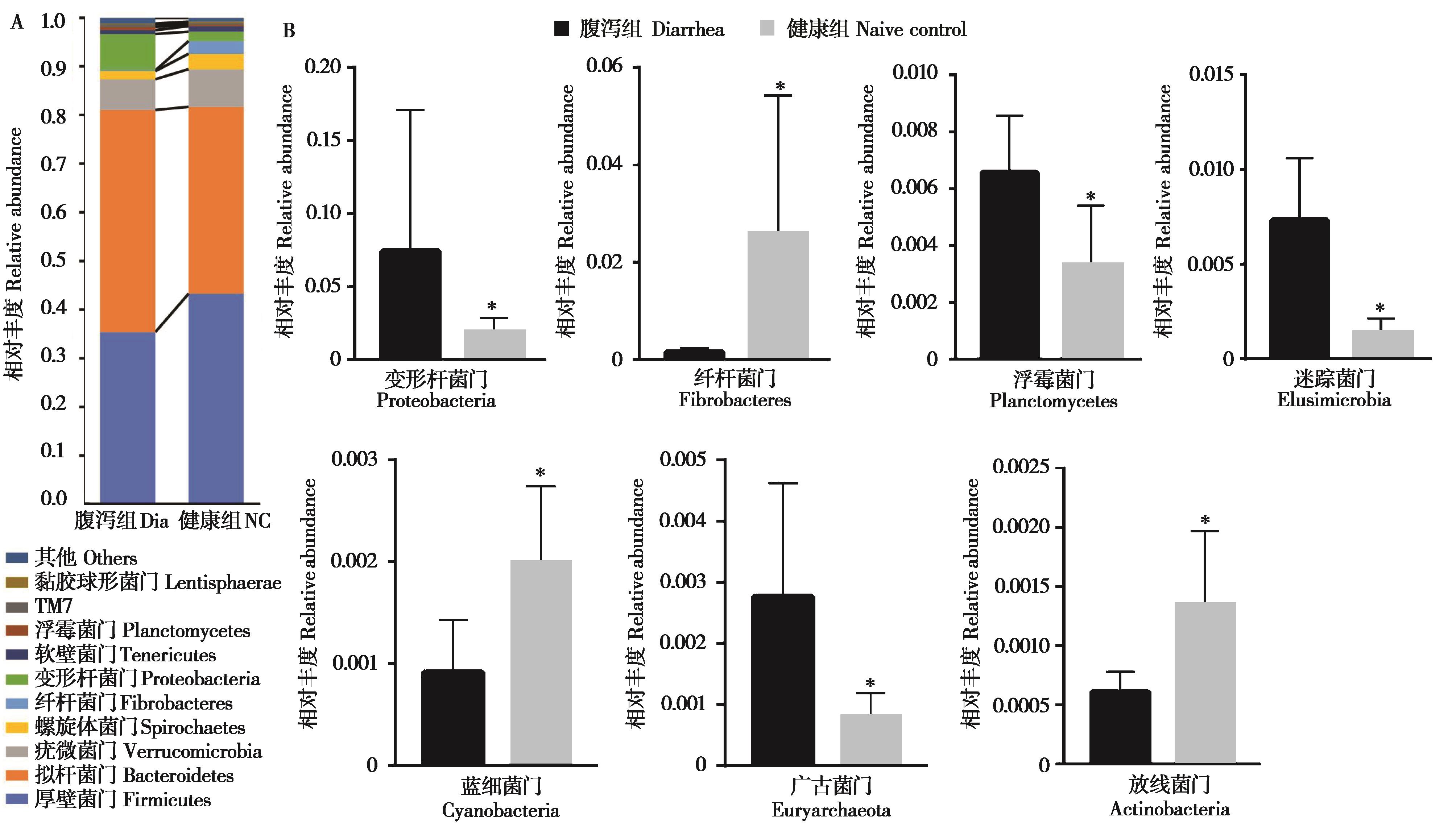
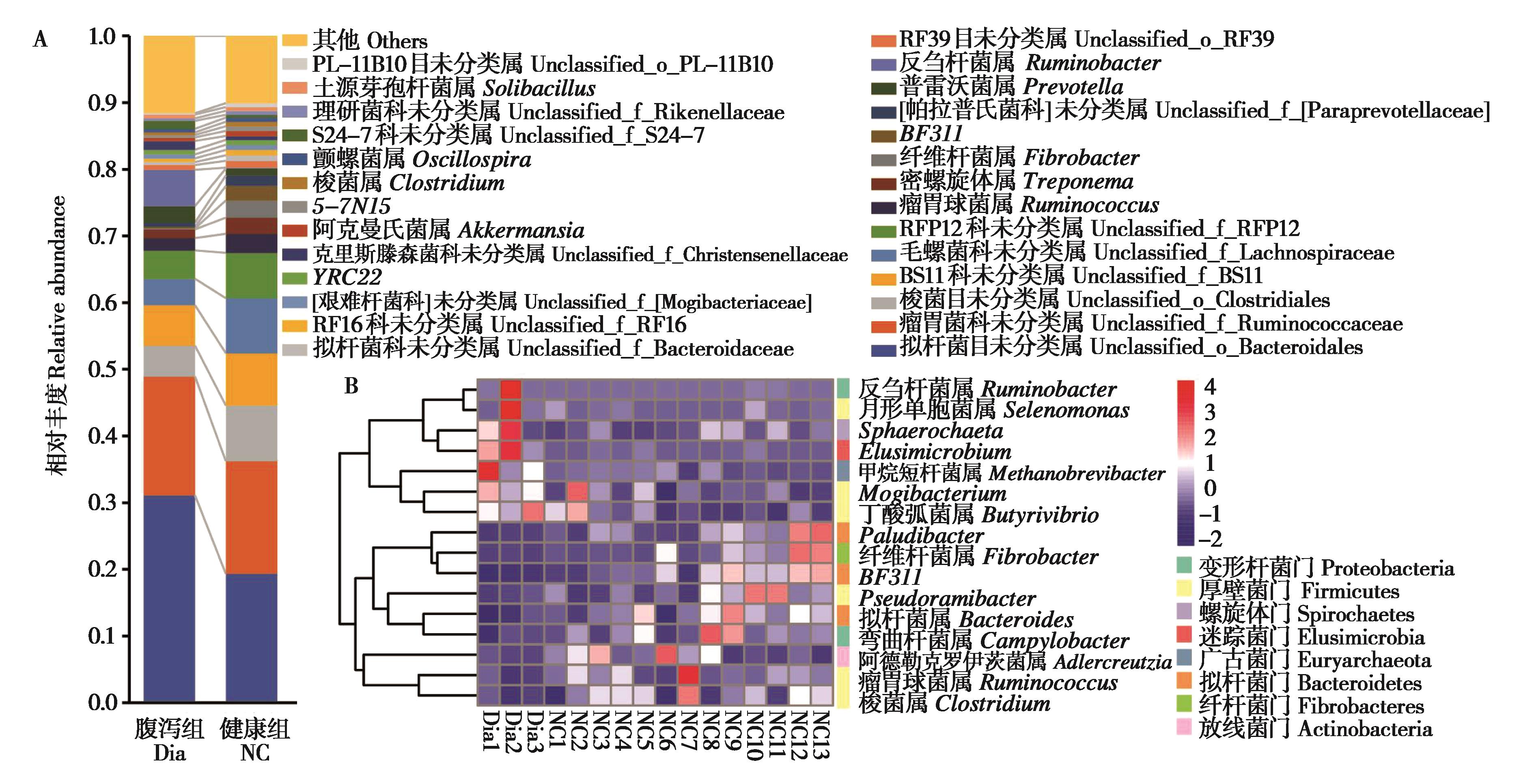
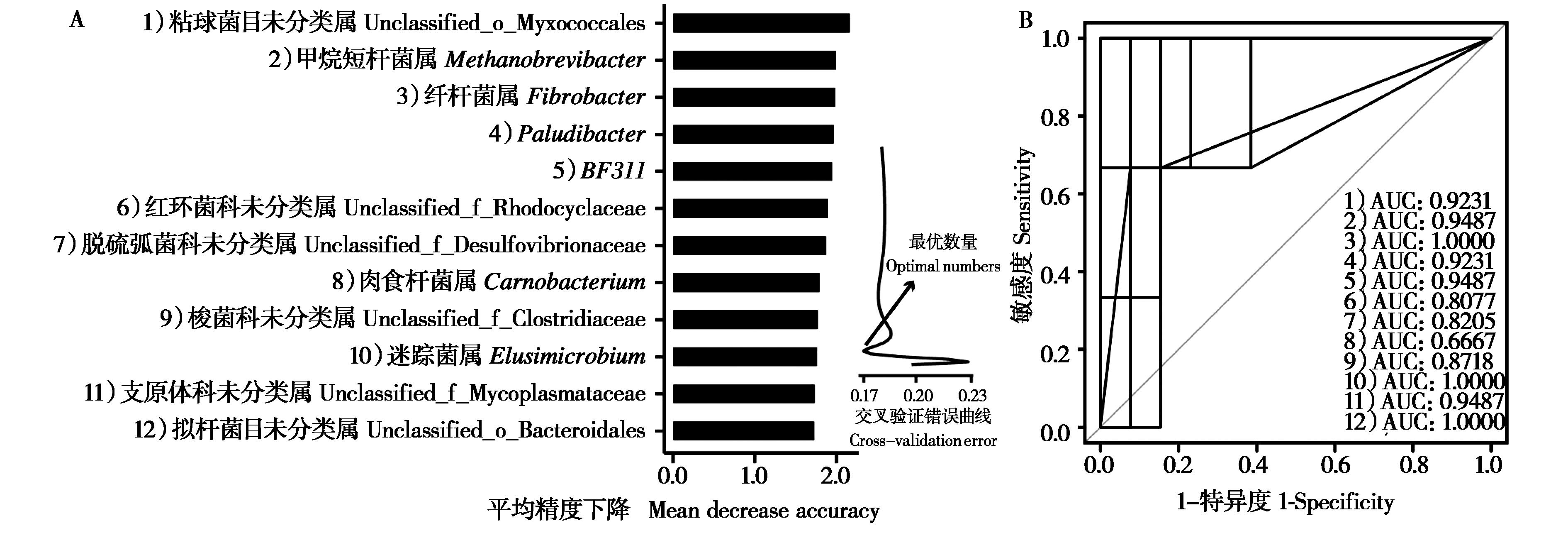
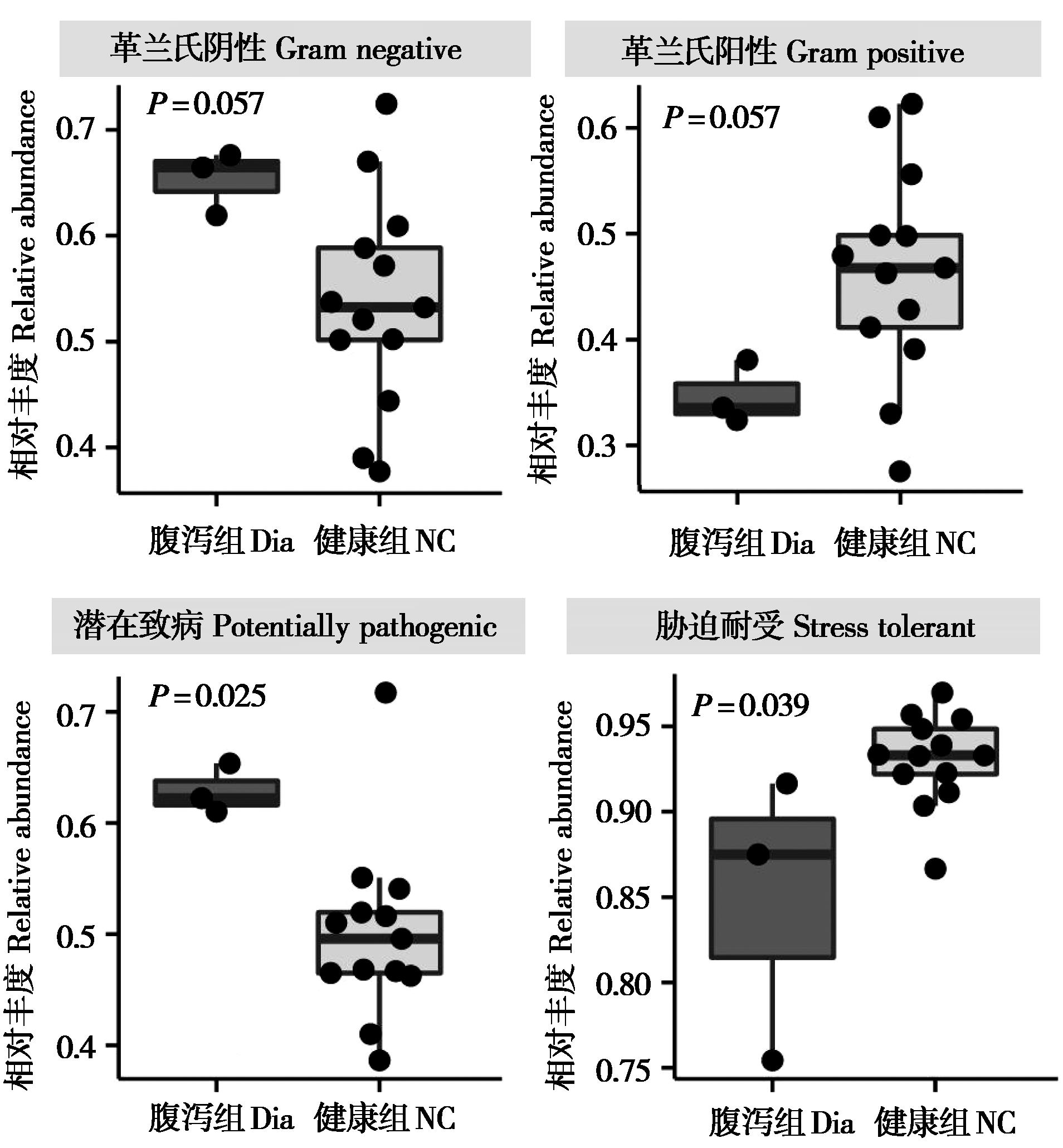
|
Al Jassim R A, Andrews F M. 2009. The bacterial community of the horse gastrointestinal tract and its relation to fermentative acidosis, laminitis, colic, and stomach ulcers. Veterinary Clinics of North America: Equine Practice, 25 (2): 199-215.
|
|
Chen S B, Ouyang Z Y, Xu W H, Xiao Y. 2010. A review of beta diversity studies. Biodiversity Science, 18 (4): 323-335. (in Chinese)
|
|
Chung W, Walker A W, Bosscher D, Garcia‑Campayo V, Wagner J, Parkhill J, Duncan S H, Flint H J. 2020. Relative abundance of the prevotella genus within the human gut microbiota of elderly volunteers determines the inter‑individual responses to dietary supplementation with wheat bran arabinoxylan-oligosaccharides. BMC Microbiology, 20 (1): 283.
|
|
Costa M C, Weese J S. 2018. Understanding the intestinal microbiome in health and disease. Veterinary Clinics of North America: Equine Practice, 34 (1): 1-12.
|
|
Costa M C, Arroyo L G, Allen‑Vercoe E, Stämpfli H R, Kim P T, Sturgeon A, Weese J S. 2012. Comparison of the fecal microbiota of healthy horses and horses with colitis by high throughput sequencing of the V3-V5 region of the 16S rRNA gene. PLoS ONE, 7 (7): e41484.
|
|
Dong Z G, Liu Y B, Pan H W, Wang H C, Wang X, Xu X F, Xiao K, Liu M, Xu Z Y, Li L B, Zhang Y. 2020. The effects of high‑salt gastric intake on the composition of the intestinal microbiota in Wistar rats.Medical Science Monitor: International Medical Journal of Experimental and Clinical Research, 26: e922160.
|
|
Drancourt M, Prebet T, Aghnatios R, Edouard S, Cayrou C, Henry M, Blaise D, Raoult D. 2014. Planctomycetes DNA in febrile aplastic patients with leukemia, rash, diarrhea, and micronodular pneumonia. Journal of Clinical Microbiology, 52 (9): 3453-3455.
|
|
Fassarella M, Blaak E E, Penders J, Nauta A, Smidt H, Zoetendal E G. 2021. Gut microbiome stability and resilience: elucidating the response to perturbations in order to modulate gut health. Gut, 70 (3): 595-605.
|
|
Garber A, Hastie P, Murray J A. 2020. Factors influencing equine gut microbiota: current knowledge. Journal of Equine Veterinary Science, 88: 102943.
|
|
Biomarkers Definitions Working Group. 2001. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clinical Pharmacology and Therapeutics, 69 (3): 89-95.
|
|
Herlemann D P, Labrenz M, Jürgens K, Bertilsson S, Waniek J J, Andersson A F. 2011. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. The ISME Journal, 5 (10): 1571-1579.
|
|
Jiang M. 2014. Analysis of difference in intestinal flora of Uygur and Han ethnic Chinese patients with ulcerative colitis. Master thesis. Urumqi: Xinjiang Medical University. (in Chinese)
|
|
Kameoka S, Motooka D, Watanabe S, Kubo R, Jung N, Midorikawa Y, Shinozaki N O, Sawai Y, Takeda A K, Nakamura S. 2021. Benchmark of 16S rRNA gene amplicon sequencing using Japanese gut microbiome data from the V1-V2 and V3-V4 primer sets. BMC Genomics, 22 (1): 527.
|
|
Kovatcheva‑Datchary P, Nilsson A, Akrami R, Lee Y S, De Vadder F, Arora T, Hallen A, Martens E, Björck I, Bäckhed F. 2015.Dietary fiber‑induced improvement in glucose metabolism is associated with increased abundance of Prevotella . Cell Metabolism, 22 (6): 971-982.
|
|
Li D L, Sun Y, Xu F R. 2021. Differences in faeces microbiome composition and characteristics between two populations of Ptyas dhumnades . Chinese Journal of Zoology, 56 (5): 696-706. (in Chinese)
|
|
Li L. 2020. Effect of different levels of tannin in diets on ruminal bacteria number and methanogens diversity in sheep. Master thesis. Hohhot: Inner Mongolia Agricultural University. (in Chinese)
|
|
Liu B D, Lin W F, Chen S J, Xiang T, Yang Y F, Yin Y L, Xu G H, Liu Z H, Liu L, Pan J Y, Xie L W. 2019. Gut microbiota as an objective measurement for auxiliary diagnosis of insomnia disorder. Frontiers in Microbiology, 10: 1770.
|
|
Mei L J, Zhou J L, Su Y M, Mao K H, Wu J, Zhu C C, He L, Cui Y. 2021. Gut microbiota composition and functional prediction in diarrhea-predominant irritable bowel syndrome. BMC Gastroenterol, 21 (1): 105.
|
|
Metzler B U, Mosenthin R. 2008. A review of interactions between dietary fiber and the gastrointestinal microbiota and their consequences on intestinal phosphorus metabolism in growing pigs. Asian Australasian Journal of Animal Sciences, 21: 603-615.
|
|
Moreno‑Indias I, Sánchez‑Alcoholado L, Pérez‑Martínez P, Andrés‐Lacueva C, Cardona F, Tinahones F, Queipo‑Ortuño M I. 2016. Red wine polyphenols modulate fecal microbiota and reduce markers of the metabolic syndrome in obese patients. Food Funct, 7 (4): 1775-1787.
|
|
Netherwood T, Wood J L, Townsend H G, Mumford J A, Chanter N. 1996. Foal diarrhoea between 1991 and 1994 in the United Kingdom associated with Clostridium perfringens, rotavirus, Strongyloides westeri and Cryptosporidium spp. Epidemiology and Infection, 117 (2): 375-383.
|
|
Oliver‑Espinosa O. 2018. Foal diarrhea: established and postulated causes, prevention, diagnostics, and treatments. Veterinary Clinics of North America: Equine Practice, 34 (1): 55-68.
|
|
Pandit R J, Hinsu A T, Patel S H, Jakhesara S J, Koringa P G, Bruno F, Psifidi A, Shah S V, Joshi C G. 2018. Microbiota composition, gene pool and its expression in Gir cattle (Bos indicus) rumen under different forage diets using metagenomic and metatranscriptomic approaches. Systematic and Applied Microbiology, 41 (4): 374-385.
|
|
Rodriguez C, Taminiau B, Brévers B, Avesani V, Van Broeck J, Leroux A, Gallot M, Bruwier A, Amory H, Delmée M, Daube G. 2015. Faecal microbiota characterisation of horses using 16 rDNA barcoded pyrosequencing, and carriage rate of clostridium difficile at hospital admission. BMC Microbiology, 15: 181.
|
|
Schoster A, Staempfli H R, Guardabassi L G, Jalali M, Weese J S. 2017. Comparison of the fecal bacterial microbiota of healthy and diarrheic foals at two and four weeks of life. BMC Veterinary Research, 13 (1): 144.
|
|
Shaw S D, Stämpfli H. 2018. Diagnosis and treatment of undifferentiated and infectious acute diarrhea in the adult horse. Veterinary Clinics of North America: Equine Practice, 34 (1): 39-53.
|
|
Shin N R, Whon T W, Bae J W. 2015. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends in Biotechnology, 33 (9): 496-503.
|
|
Stamps B W, Lyon W J, Irvin A P, Kelley‑Loughnane N, Goodson M S. 2020. A pilot study of the effect of deployment on the gut microbiome and Traveler’s Diarrhea susceptibility. Frontiers Cellular and Infection Microbiology, 10: 589297.
|
|
Stern E K, Brenner D M. 2018. Gut microbiota‑based therapies for irritable bowel syndrome. Clinical and Translational Gastroenterology, 9 (2): e134.
|
|
Wang B, Deng B, Yong F, Zhou H X, Qu C P, Zhou Z Y. 2020. Comparison of the fecal microbiomes of healthy and diarrheic captive wild boar. Microbial Pathogensis, 147: 104377.
|
|
Wang X Q, Zhang Z C, Wang X P, Bao Q, Wang R J, Duan Z Y. 2021. The impact of host genotype, intestinal sites and probiotics supplementation on the gut microbiota composition and diversity in sheep. Biology (Basel), 10 (8): 769.
|
|
Ward T, Larson J, Meulemans J, Hillmann B, Lynch J, Sidiropoulos D, Spear J R, Caporaso G, Blekhman R, Knight R, Fink R, Knights D. 2017. BugBase predicts organism-level microbiome phenotypes. bioRxiv: 133462.
|
|
Xi L, Song Y M, Han J C, Qin X X. 2021. Microbiome analysis reveals the significant changes in gut microbiota of diarrheic Baer’s Pochards (Aythya baeri). Microbial Pathogenesis, 157: 105015.
|
|
Xie H S, Huan L, Merritt A M, Ott E A. 1997. Equine chronic diarrhea: traditional chinese veterinary medicine review. Journal of Equine Veterinary Science, 17 (12): 667-674.
|
|
Xia Y, He X W, Wen X H. 2018. Mathematic methods for the evaluation of microbial diversity and their applications in the research on wastewater treatment systems. Microbiology China, 45 (8): 1778-1786. (in Chinese)
|
|
Yang L N, Bian G R, Zhu W Y. 2014. Interactions between the monogastric animal gut microbiota and the intestinal immune function- A review. Acta Microbiologica Sinica, 54 (5): 480-486. (in Chinese)
|
|
Zeng Q, Li D F, He Y, Li Y H, Yang Z Y, Zhao X L, Liu Y H, Wang Y, Sun J, Feng X, Wang F, Chen J X, Zheng Y J, Yang Y Y, Sun X L, Xu X M, Wang D X, Kenney T, Jiang Y Q, Gu H, Li Y L, Zhou K, Li S C, Dai W K. 2019. Discrepant gut microbiota markers for the classification of obesity‑related metabolic abnormalities. Scientific Reports, 9 (1): 13424.
|
|
Zhai Q X, Tian F W, Wang G, Chen W. 2013. Progress in research on the role of intestinal microbiota in human health. Food Science, 34 (15): 337-341. (in Chinese)
|
|
Zhu H, Zeng D, Wang Q, Wang N, Zeng B, Niu L L, Ni X Q. 2018. Diarrhea‑associated intestinal microbiota in captive Sichuan golden snub‑nosed monkeys (Rhinopithecus roxellana). Microbes and Environments, 33 (3): 249-256.
|
|
DB 63/T962-2011. 2011. 柴达木马. 青海省: 青海省质量技术监督局.
|
|
陈圣宾, 欧阳志云, 徐卫华, 肖燚. 2010. Beta多样性研究进展. 生物多样性, 18 (4): 323-335.
|
|
翟齐啸, 田丰伟, 王刚, 陈卫. 2013. 肠道微生物与人体健康的研究进展. 食品科学, 34 (15): 337-341.
|
|
蒋曼. 2014. 新疆维吾尔族与汉族溃疡性结肠炎患者肠道菌群差异性比较. 乌鲁木齐: 新疆医科大学硕士学位论文.
|
|
李栋梁, 孙越, 徐发荣. 2021. 乌梢蛇粪便微生物群组成和特征的种群间差异. 动物学杂志, 56 (5): 696-706.
|
|
丽丽. 2020. 饲粮中不同水平单宁对绵羊瘤胃细菌菌群数量及产甲烷菌多样性的影响. 呼和浩特: 内蒙古农业大学硕士学位论文.
|
|
夏瑜, 何绪文, 文湘华. 2018. 微生物群落多样性数学表征方法及其在污水处理系统研究中的应用. 微生物学通报, 45 (8): 1778-1786.
|
|
杨利娜, 边高瑞, 朱伟云. 2014. 单胃动物肠道微生物菌群与肠道免疫功能的相互作用. 微生物学报, 54 (5): 480-486.
|


 )
)
 青公网安备 63010402000199号 青ICP备05000010号-2
青公网安备 63010402000199号 青ICP备05000010号-2